
有道开口读最新版
627.12MB · 2025-12-23

蛋白质序列对齐 (比较蛋白质序列的相似性) 是现代生物学和医学的基础。它通过重建进化关系 (技术上称为 homology inference) 来照亮基因功能,为药物开发提供信息。当科学家发现或设计一种新蛋白质时,他们可以将其与已知的蛋白质序列进行比对,以推断其结构和功能。
这种同源性搜索可以揭示有前景的药物点 (例如,通过比较病原体蛋白质与人类蛋白质) 或查明致病突变 (通过比较患者的蛋白质与健康蛋白质) 。然而,基因组和宏基因组数据的快速扩展现在给传统的对齐工具带来了压力。
本文将探讨蛋白质对齐方面的最新进展如何通过使用 GPU 优化的对齐,以前所未有的速度增强 AI 驱动的药物发现、结构预测和蛋白质设计,从而加速蛋白质科学。
序列对齐可能听起来技术含量很高,但其重要性显而易见:科学家可以比较蛋白质序列以找到相似性。相似序列通常意味着相似的功能或结构特征。这是同源性推理的基础:如果蛋白质 A 与蛋白质 B 相似,则它们可能具有生物学作用。
蛋白质序列对齐通过识别保守区域、预测蛋白质功能和检测可能导致疾病的不太可能的突变,对功能注释、进化研究、疾病研究和药物研发至关重要。在序列对齐中编码的进化信息还可以指导药物点的选择和优化。

人类蛋白质组表现出极大的复杂性。典型的乳腺细胞包含大约 10 billion 个蛋白质分子,每个细胞的丰度动态范围为 106。像 plasma 这样的体液在丰富程度最高和最少的蛋白质之间表现出 1010 倍的差异。这种复杂性给全面的蛋白质组分析带来了重大挑战。
绘制蛋白质相互作用图更加令人望而生畏。虽然人类蛋白质组中存在近 200 million 种可能的成对相互作用,但只有约 53,000 种经过实验证实,这使得这种映射类似于在大海捞针。
从头设计蛋白质是这些挑战中计算复杂度最高的,涉及探索天文搜索空间 (N 长度蛋白质的 20N 序列) ,并解决折叠和功能优化中的 NP 难题。AI 和自动化的进步显著提高了成功率 (在某些设计类别中,成功率从约 10% 提高到约 30 – 50%) ,但实验验证仍然需要大量资源,通常需要进行迭代测试和优化。虽然近期的突破加速了进展,但这些基本问题仍处于结构生物学的前沿。
生物信息学家已经开发出越来越高效的算法来加速序列对齐。BLAST 在 20 世纪 90 年代彻底改变了搜索速度,但却难以应对不断增长的数据,因此在 2010 年代开发出了 DIAMOND 和 MMseqs2。
MMseqs2 的灵敏度优于 PSI-BLAST,运行速度提高了 400 多倍。在三次迭代的配置文件搜索中,MMseqs2 的速度比 PSI-BLAST 快 433 倍,同时还表现出更高的灵敏度。
MMseqs2 和 DIAMOND 现已广泛应用于基因组注释和药物研发,取代了曾经需要数周计算才能完成的工作流中的 BLAST。然而,随着数据量的持续爆炸,即使是基于 CPU 的最快工具也面临着限制,这促使人们转向 GPU 加速。尽管 AI 取得了进步,但蛋白质对齐仍然至关重要。AlphaFold2 等深度学习模型依靠多序列对齐 (MSA) 来预测蛋白质结构,从而证明快速且可扩展的序列搜索方法的持久重要性 (表 1) 。
| MMseqs2 用例 | 示例 | 参考资料 |
| 扩展和级联 MSA 搜索以预测蛋白质结构 |
| 使用 AlphaFold 实现高度准确的蛋白质结构预测 ColabFold:让所有人都能进行蛋白质折叠使用三轨神经网络准确预测蛋白质结构和相互作用 OpenFold:重新训练 AlphaFold2 让人们对其学习机制和泛化能力有了新的认识 |
| 过滤冗余或同源序列 | ADOPT:识别内在无序蛋白质区域 | ADOPT:通过深度双向Transformers预测内部蛋白质疾病 |
| 用于蛋白质相互作用分析的Clustering | 感知 – PPI | SENSE-PPI 以基因组尺度重建物种内部、之间和之间的相互作用组 |
在基于深度学习的工作流程中,更快的 MSA 具有巨大的潜在优势。MSA 搜索的计算成本高昂,通常会占用推理和训练时间。例如,AlphaFold2 和 OpenFold 的推理主要由 MSA 搜索进行 (约占总时间的 70 – 90%) ,与单个计算作业相比,将 MSA 对齐和折叠算法拆分成两个作业可节省 AlphaFold 57% 的成本,而 OpenFold 可节省 51% 的成本。
MMseqs2-GPU 利用特定于 GPU 的新型加速在 CUDA 上解锁多个序列对齐。开发 MMseqs2-GPU 的联合研究团队由首尔大学、Johannes Gutenberg University Mainz 和 NVIDIA 的研究人员领导。该团队创建了一种针对 GPU 优化的新型“无缝”过滤算法,以取代基于 CPU k-mer 的预过滤器。简单来说,GPU 版本并不像在 CPU 上扫描 BLAST 和 MMseqs2 那样扫描短匹配子串 (k-mers) ,而是使用高度并行算法,直接在序列中对对齐进行评分,而没有间隙 (允许不匹配,但没有插入/ 删除) 。
这种使用 CUDA 实施的方法经过专门定制,可避免 memory bottlenecks,同时使数千个 GPU 核心保持忙碌。在此快速预过滤器找到有前景的匹配后,GPU 还可以针对这些命中执行更精确的 gapped alignment(使用优化的 Smith-Waterman 算法)。
为了全面了解这一进步,MMseqs2 GPU 技术报告中的主要性能比较包括以下内容:
作为 MMseqs2-GPU 在药物研发工作流程中解决现实问题的示例,以下展示了序列对齐在 AI 驱动的目标结构预测中的作用。此处,MMSeqs2-GPU (MSA-Search) 和 OpenFold 模型均被打包为容器化的 NVIDIA NIM 微服务(图 2)。
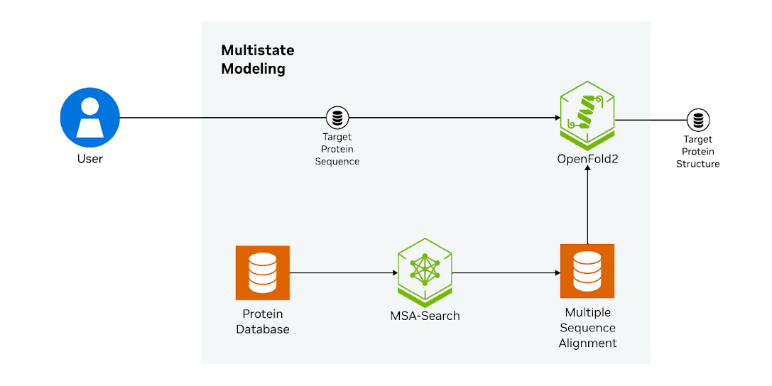
例如,MSA-Search NIM 和 OpenFold2 NIM 可以一起使用,对目标蛋白质的多个构象状态进行建模。工作流程涉及以下步骤:
以下代码以人类 hCLpP 蛋白质为例,说明了如何做到这一点:
"""Example: model two conformations of human ClpP (hClpP)"""import os, json, requests, pathlib# ------------------------------------------------------------------# Config# ------------------------------------------------------------------API_KEY = os.environ["NIM_API_KEY"]HEADERS = {"Authorization": f"Bearer {API_KEY}", "Content-Type": "application/json"}MSA_URL = "http://localhost:8000/biology/colabfold/msa-search/predict"OF2_URL = "http://localhost:8000/biology/openfold/openfold2/predict-structure-from-msa-and-template"SEQ = """>hClpP_HUMANMARGKIIGELASKKKVEAMAAKLAEAG... (FASTA truncated for clarity)"""# ------------------------------------------------------------------# 1) Run GPU-MMseqs2 via MSA-Search NIM# ------------------------------------------------------------------msa_payload = { "sequence": SEQ, "databases": ["uniref90-2024_02", "pdb70"], "return_templates": True # tells the service to emit an HHR-formatted hit list}msa_resp = requests.post(MSA_URL, headers=HEADERS, data=json.dumps(msa_payload), timeout=900)msa_resp.raise_for_status()msa = msa_resp.json()a3m_alignment = msa["alignments"]["uniref90-2024_02"]["a3m"]["alignment"]# Helper: pick two PDB templates that represent distinct states.ACTIVE_PDB = "7DKF" # hClpP active/openINACTIVE_PDB = "7D7G" # hClpP inactive/closedhhr_all = msa["templates"]["pdb70"]["hhr"]["alignment"] # full HHR textdef slice_hhr_for(pdb_id: str, hhr_text: str) -> str: """Return an HHR minimal block for a single template PDB hit.""" keep = [] write = False for line in hhr_text.splitlines(): if line.startswith(">PDBID:") and pdb_id in line: write = True elif line.startswith(">PDBID:") and write: break if write: keep.append(line) return "n".join(keep)hhr_active = slice_hhr_for(ACTIVE_PDB, hhr_all)hhr_inactive = slice_hhr_for(INACTIVE_PDB, hhr_all)# ------------------------------------------------------------------# 2) Predict *active* conformation with OpenFold 2# ------------------------------------------------------------------def run_openfold2(hhr_block: str, tag: str) -> pathlib.Path: payload = { "sequence": SEQ, "alignments": { "uniref90-2024_02": { "a3m": {"alignment": a3m_alignment, "format": "a3m"} } }, "templates": { "pdb70": { "hhr": {"alignment": hhr_block, "format": "hhr"} } }, "selected_models": [1] # run a single model for speed; omit for ensemble } r = requests.post(OF2_URL, headers=HEADERS, data=json.dumps(payload), timeout=1800) r.raise_for_status() pdb_text = r.json()["predictions"][0]["structure"] out = pathlib.Path(f"hClpP_{tag}.pdb") out.write_text(pdb_text) print(f"Wrote {out}") return outactive_pdb = run_openfold2(hhr_active, "active")inactive_pdb = run_openfold2(hhr_inactive, "inactive")从 BLAST 到 MMseqs2-GPU,序列对齐方面的进步彻底改变了蛋白质科学,使人们能够更快地了解功能、进化和药物研发。
此工具已用于合成数据集生成(AI 模型开发生命周期的重要组成部分),以及用于测试时扩展和实时预测的加速推理。MMseqs2-GPU 已在业内得到广泛采用,包括 Basecamp Research、VantAI 和 Iambic Therapeutics 等领先公司。
随着 AI 驱动的模型将对齐集成到预测工作流程中,GPU 加速正在重新定义分子研究。AI、HPC 和生物信息学的融合有望实现更大的突破,加速医学和生物技术的发现。详细了解用于生成式蛋白质结合剂设计的 NVIDIA BioNeMo Blueprint。
试用 MMSeqs2-GPU 作为 NVIDIA MSA-Search NIM,并试用 OpenFold 作为 NVIDIA OpenFold2 NIM。

